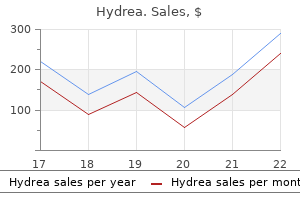
500 mg hydrea quality
It is usually associated with pores and skin modifications and abnormalities of the decrease extremities symptoms gerd trusted hydrea 500 mg. A chordoma is a neoplasm that arises from notochordal rests and is found especially within the sacrococcygeal area medicine 122 500 mg hydrea generic mastercard. Sacrococcygeal teratoma is a neoplasm composed of multiple embryonic tissues that can undergo malignant transformation. The neural tube becomes the central nervous system (the brain and spinal cord) and the neural crest develops into most of the peripheral nervous system. The neural tube varieties from the folding of the neural groove and begins at the brain/spinal cord junction. Neural crest cells begin their migration from neural fold tissue just after neural tube closure happens within the spinal areas. Neural crest cells migrate both beneath the surface ectoderm or between the neural tube and the somite. Neural crest cells from the pia matter, the spinal ganglia, and the sympathetic trunks and ganglia. As the neural tube closes, the dorsal area, called the alar laminae is separated from the ventral basal laminae by a shallow groove - the sulcus limitans. A thin bridge of tissue persists to connect the 2 halves of the alar and basal laminae named the roof and ground plates. The alar plate develops into the sensory pathways (dorsal columns) and the basal plate develops into the motor pathways (ventral horns). The spinal twine extends the complete size of the vertebral column in the course of the embryonic interval. During fetal development, the vertebral column grows more quickly than the spinal cord. Coupled with some lack of caudal spinal cord tissue, the caudal tip of the spinal twine ends at the 2nd or third lumbar vertebra in newborns. In the adult, the spinal cord terminates on the inferior portion of the primary lumbar vertebra. Myelination of peripheral nerve axons begins in the fetal period and continues in the course of the first year after start. Schwann cells myelinate peripheral nerves, whereas oligodendrocytes myelinate the axons throughout the spinal wire. The transcription factor genes Pax-3 and Gli-3 are needed for neural tube closure (92). Myeloschisis is a uncommon condition during which the neural groove fails to form a neural tube. Myelomeningocele has uncovered neural tissue that has herniated into the dysraphic area of the spine. Meningocele, during which the neural elements stay in their regular location, might be a major defect of the vertebral column improvement, quite than a primary neural defect. Ten % of people have spina bifida occulta during which the vertebral neural arch fails to absolutely develop and fuse, normally at L5 or S1. Hepatocyte nuclear factor-3 seems to regulate sonic hedgehog (an important axis specifying gene in the limb as well), which may induce ventral buildings (90). Dorsal buildings can develop without the notochord, but specific molecules may be essential for complete dorsal specification. Illustration of the place of the spinal wire and meninges in relation to the vertebral column at (A) 8 weeks, (B) 24 weeks, (C) start, and (D) grownup. Regardless of the animal species or the bone thought of, bone is all the time a two-phase composite substance made up of two very different supplies. The two main parts of bone are the organic matrix, or osteoid, and the inorganic matrix. Various calcium salts, primarily hydroxyapatite, are deposited in crystals within and between the matrix. These inorganic crystals give bone its rigidity, hardness, and strength to compression. Connective tissue, cartilage, and bone all differentiate from that type of diffuse mesoderm known as mesenchyme. In early embryos, the mesenchyme acts as a unspecialized "packing" materials however quickly differentiates into varied tissues and organs. There are two mechanisms of bone formation (osteogenesis), and each involve the transformation of a preexisting connective tissue into bone tissue. The transformation of fibrous primitive connective tissue into bone is called intramembranous ossification. Except the clavicle and the flat bones of the skull, all bones of the appendicular and axial skeleton type by endochondral ossification. During intramembranous ossification, the mesenchymal cells proliferate and condense into packed nodules. Some of those cells differentiate into capillaries and others change their form to turn into osteoblasts. These cells are capable of secreting osteoid, the natural extracellular matrix that subsequently will turn into mineralized. High ranges of alkaline phosphatase and the looks of matrix vesicles mark the graduation of ossification. The cells will eventually be surrounded by calcified matrix and turn out to be osteocytes. Intramembranous ossification occurs by mesenchymal cells derived from the neural crest that interact with the extracellular matrix secreted by the epithelia cells arising from the pinnacle. The mechanism responsible for the conversion of mesenchymal cells to bone is still unknown. By far, the most typical mechanism of ossification is cartilaginous (or endochondral). Mesenchyme cells condense and proliferate however as a substitute of turning into osteoblasts, like in intramembranous ossification, they turn out to be chondroblasts. Soon after the cartilaginous mannequin is shaped, the cells in the middle turn out to be hypertrophic and secrete a matrix that may subsequently be invaded by capillaries. As this matrix is degraded and the chondrocytes die, osteoblasts carried by the blood vessels start to secrete bone matrix. This process seems to be dependent on the mineralization of the extracellular matrix. Interestingly, a special, condensed mesenchymal tissue, the perichondrium, surrounds the cartilage model. This tissue is basically the identical as that surrounding the intramembranous centers of ossification, but within the perichondrium the osteoprogenitor cells stay dormant for a time, while the cartilage mannequin is enlarged by the chondrocytes. Ossification begins on the major center, throughout the shaft, and proceeds outward from the medullary cavity and inward from the periosteum in a repetitive sequence. Second, the developing bone continues to enlarge by both interstitial and appositional growth.
Diseases
- Arroyo Garcia Cimadevilla syndrome
- Bassoe syndrome
- Hemochromatosis
- Oculo tricho anal syndrome
- Hunter Carpenter Mcdonald syndrome
- Spontaneous periodic hypothermia
- CDG syndrome type 3
Hydrea 500 mg order without prescription
This repeats can affect the protein-coding or no-coding regions and result in translation of a series of uninterrupted residues that have an result on protein function medications with acetaminophen generic hydrea 500 mg online. Importantly treatment diabetic neuropathy hydrea 500 mg cheap with mastercard, trinucleotide repeat disorders usually present genetic anticipation, where their severity will increase with each successive generation that inherits them, likely defined by the addition of additional repeats in the gene of the progeny of affected people. Mosaicism refers to mutations that result in cell clones which are genetically totally different from the unique zygote. Because the members of a clone are likely to stay near each other during growth, an observable outcome of a somatic mutation is often a patch of phenotypically mutant cells referred to as a mutant sector. The earlier the mutation event in improvement, the bigger the mutant sector might be. Like each other organ system, the skeleton has specific developmental and useful traits that outline its identification in biologic and pathologic terms. For normal skeletogenesis to happen, the coordination of temporal and spatial gene expression patterns is an important prerequisite. The vertebrate skeleton is fashioned by mesenchymal cells condensing into tissue components outlining the pattern of future bones (the patterning phase). The cartilage anlagen will be replaced by bone and bone marrow in a course of referred to as endochondral ossification. Mutations in early patterning genes cause problems referred to as dysostoses: these affect only specific skeletal components, leaving the the rest of the skeleton largely unaffected. In contrast, mutations in genes that are involved primarily in cell differentiation cause disorders referred to as osteochondrodysplasias, which have an result on the development and progress of most skeletal elements in a generalized trend. Many genes have necessary capabilities in each of these processes so that some inherited issues can show options of both dysostoses and osteochondrodysplasias. Genes used during skeletal improvement may also be essential in different organs, so when mutated, the resulting skeletal defects are part of a syndrome. There have been large advances in the past 50 years figuring out the causative genetic defect for lots of the problems that are handled by pediatric orthopaedists. At the chromosomal degree, the area of the chromosome containing the illness gene may be revealed by cytogenetic evaluation. Translocations might disrupt a gene and can subsequently produce the disease, and a microdeletion might indicate lack of contiguous genes. Translocations, which are widespread in many tumors, might interrupt and inactivate a gene or could end result within the fusion of two genes, which then produce a model new fusion protein. The research of contiguous gene deletion syndromes has enabled researchers to affiliate these genes with specific phenotypes. In some illnesses, candidate genes are selected and tested for their affiliation with the illness. For example, the type I collagen genes had been the candidate genes in osteogenesis imperfecta because type I collagen is found in the entire major tissues affected by the disease. Mutational analyses can then be undertaken to determine the genotypes and the genotype/phenotype relations. Genetic linkage analysis is the standard approach used for figuring out a disease gene in people when no probably candidate genes can be postulated or the place candidate gene screening has not revealed any anomalies. Linkage evaluation is based on figuring out whether genetic markers or polymorphisms, either within or flanking the candidate gene, are coinherited with the illness phenotype in families. Large families are usually needed for such studies, and cautious analysis is needed in classifying individuals as phenotypically affected or unaffected. Phenotypic ascertainment may be easy, as in classical Ehlers-Danlos syndrome kind I, during which signs include pores and skin scars, skin laxity, and generalized joint instability. The syndrome is fully penetrant, in that all individuals bearing the mutant allele show the clinical phenotype. The pores and skin and joints are clearly irregular in any respect ages and in each sexes although the severity of the skin scarring worsens with age. In distinction, it might be troublesome in the case of other genetic problems to clinically decide whether or not asymptomatic people bear the mutant allele or not. This problem could also be as a outcome of low penetrance, variable expressivity, age, and gender. Such difficulties are prone to account for the lack of progress in figuring out genes for widespread situations corresponding to idiopathic scoliosis, clubfoot, and developmental dysplasia of the hip (14, 15). Also, newer technology sequencing techniques are in development that makes it potential to do whole genome sequencing, providing a high-throughput technique to establish genetic abnormalities (16). Mouse genetic studies are an integral part of the successful identification of disease genes and their function in humans. There are many examples of mouse fashions of human illnesses and even fruit fly models of human ailments. Some of these fashions have been the outcomes of spontaneous mutations, whereas others were produced by focused mutations or inactivation of genes of curiosity. Genetic studies of mice are useful in establishing whether or not a putative missense mutation identified in people is a cause of a given phenotype. Significant progress is being made in figuring out the genes involved in multigene or multifactorial disorders of the musculoskeletal system, for example, in degenerative arthritis, intervertebral disk disease, and osteoporosis. The latter studies present new insights into the etiologies of those issues and present that some of the multifactorial issues are a half of larger disease households. It is most likely going that shut links might be established between many of the rare single-gene disorders and the widespread multigene issues of the musculoskeletal system. Broadly outlined, birth defects or congenital abnormalities happen in 6% of all reside births. Combined genetic predisposition with environmental elements causes the remaining 20% to 25% of congenital abnormalities (7). Although individually uncommon, the completely different varieties add to produce a major variety of affected people, with significant morbidity. Clinical manifestations vary from neonatal lethality to congenital malformations of the spine and limbs, to solely gentle growth retardation. Importantly, secondary issues similar to early degenerative joint illness and extra-skeletal organ involvement add to the burden of the disease. Their clinical variety makes these issues typically troublesome to diagnose, and plenty of attempts have been made to delineate single entities or groups of ailments to facilitate the analysis. Traditionally, skeletal problems have been subdivided into dysostoses, outlined as malformations of individual bones or groups of bones, and osteochondrodysplasias, outlined as developmental issues of cartilage and bone. The standards used for their distinction has been primarily based on a mixture of scientific, radiographic, morphologic, and, in a number of cases, biochemical characteristics. The modes of genetic inheritance and extra skeletal abnormalities have also been used. The International Working Group on the Classification of Constitutional Disorders of Bone updated the classification in 2001 (17).
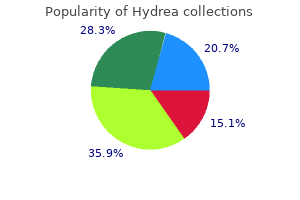
Purchase 500 mg hydrea otc
Gene expression profiling reveals signatures characterizing histologic subtypes of hepatoblastoma and global deregulation in cell growth and survival pathways 8h9 treatment order hydrea 500 mg. Epigenetic regulation of most cancers stem cells in liver cancer: current ideas and scientific implications treatment for bronchitis discount 500 mg hydrea overnight delivery. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Transcriptomic and genomic evaluation of human hepatocellular carcinomas and hepatoblastomas. Complete surgical resection is curative for children with hepatoblastoma with pure fetal histology. Factors predicting response and survival in 149 sufferers with unresectable hepatocellular carcinoma handled by combination cisplatin, interferon-alpha, doxorubicin and 5-fluorouracil chemotherapy. Thalidomide inhibits the expansion and progression of hepatic epithelioid hemangioendothelioma. Patients who present with acute hepatitis, even with delicate coagulopathy, ought to be thought of for transfer early of their course. Timely referral for transplantation must happen earlier than the anticipated progressive deterioration associated with liver illness, and before lifethreatening problems, or contraindications to transplantation, happen. There are numerous key questions that ought to be answered with each transplant evaluation. Will liver transplantation enhance each short-term and long-term survival compared with no transplantation? Is there irreversible and progressive non-hepatic illness that can negate the effects of liver transplantation on outcome? Additional goals of the analysis embrace guaranteeing that medical and surgical management is optimized and that an efficient pretransplant care plan is developed. This consists of identifying non-hepatic issues of liver disease that might adversely have an effect on the operative and postoperative consequence. Malnourished sufferers ought to have their dietary intake optimized by way of the use of nasoenteric feeding tubes and/or high-calorie formulae. Medications should be adjusted to treat encephalopathy, ascites, or different complications of liver illness as needed. Prophylaxis for bacterial peritonitis or recurrent cholangitis also needs to be administered when indicated. Immune standing associated to prior viral exposures and routine childhood immunizations must be assessed. Immunizations, particularly attenuated live viruses, and preventive dental care must also be administered if time permits. The patient and family must also be educated concerning expectations when positioned on the waiting list. The household Liver transplantation has turn into the standard of take care of end-stage liver illness in youngsters and profitable outcomes at the moment are achieved in the vast majority of transplant recipients. Progressive improvement has occurred via higher preoperative care of patients with liver disease, improved operative methods that has allowed the donor pool to increase, and improved immunosuppression methods to forestall rejection while avoiding complications of over-immunosuppression. With the growing number of liver transplant candidates, improved donor awareness and organ availability must happen. A delicate steadiness between the dangers assumed by dwelling donors and the wants of their kids have to be struck. The increasing numbers of surviving patients present distinctive challenges and issues associated to lifelong immunosuppression. The future success of pediatric liver transplantation would require appreciation of the increasingly advanced care needs of this population and a national concentrate on donor organ shortages. The main goal of a transplant analysis is to identify candidates for whom liver transplantation is the optimum treatment. It can additionally be beneficial to discuss the indications for dwelling related liver transplantation, such because the survival benefit in younger sufferers, and the danger to the donors, who are sometimes the dad and mom. Older patients must also be encouraged to ask questions and if potential ought to provide assent to the operation. It is prudent to start educating adolescent or preteenage sufferers relating to treatment adherence as well as alcohol or other substance avoidance. Indications and contraindications to liver transplantation the commonest scientific displays prompting transplant evaluation in children can be categorized as (1) cholestatic liver disease, (2) continual liver disease with extrahepatic problems, (3) metabolic disease correctable with liver replacement, (4) acute liver failure, and (5) unresectable liver tumors, together with vascular malformations which may lead to progressive coronary heart failure. Absolute contraindications to transplantation embrace (1) main extrahepatic unresectable malignancy, (2) progressive terminal non-hepatic illness, (3) uncontrolled systemic sepsis, and (4) irreversible neurologic damage. Clinical failure of the Kasai procedure may be manifest by varied combos of issues corresponding to recurrent bacterial cholangitis, progressive portal hypertension with refractory ascites or variceal bleeding, malnutrition, and/or progressive hepatic artificial failure. Approximately 50% of all infants with biliary atresia could have these problems and would require liver transplant within the first 2 years of life. Even kids with the profitable institution of biliary drainage and normalization of serum bilirubin levels post-Kasai may develop progressive cirrhosis with portal hypertension, hypersplenism, variceal hemorrhage, and ascites formation. These complications typically result in liver transplantation after the child is over 2 years of age. An additional non-hepatic complication in sufferers with biliary atresia, in addition to another type of continual liver illness, with or with out portal hypertension is hepatopulmonary syndrome, which might even happen in sufferers with stable liver artificial operate. Subtle medical adjustments similar to train intolerance, decreased energy, a decline in room air oxygen saturations, clubbing of the fingers, or spider telangiectasias ought to prompt screening for hepatopulmonary syndrome with arterial blood gasoline or echocardiography. Children with hepatopulmonary syndrome ought to bear liver transplantation to keep away from progressive hypoxia or later mounted pulmonary hypertension. Other cholestatic conditions, similar to progressive familial intrahepatic cholestasis or Alagille syndrome may trigger cirrhosis with artificial liver dysfunction and/or portal hypertension inflicting variceal bleeding. In children afflicted with these conditions, indications for transplant include intractable pruritus, xanthomata, marked osteodystrophy with recurrent fractures recalcitrant to medical remedy, or, hardly ever, hepatocellular carcinoma [5]. Hepatic-based metabolic disease A leading indication for liver transplantation in children is hepatic-based metabolic illness (see the relevant chapters for added info on these disorders). Liver substitute to right the metabolic defect must be thought-about before other organ methods are affected or the implications of the defect lead to irreversible quality-of-life compromises or complications that might prove to be contraindications to transplantation. Early transplantation permits the potential for neurologic protection and restoration with preservation of neurologic perform and quality of life. Primary liver illnesses leading to liver transplantation Cholestatic syndromes Children with biliary atresia represent approximately 50% of the pediatric liver transplant inhabitants. Portoenterostomy (the Kasai procedure) should be the first surgical intervention for all infants with biliary atresia unless the preliminary presentation is late in infancy (>120 days of age), the liver biopsy shows advanced cirrhosis, or the medical course is unfavorable. Approximately 15Ͳ0% of patients with biliary atresia may not require liver transplantation [3]. Currently, nitisinone (2-(2-nitro-4-trifluoromethylbenzoyl)-1, 3-cyclohexanedione) is taken into account to be first-line remedy for tyrosinemia. Nitisinone, through inhibition of 4-hydroxyphenylpyruvate oxidase, prevents dysplasia and hepatocellular carcinoma, thus obviating the need for liver transplantation. Neonatal hemochromatosis (also often identified as neonatal iron storage disease or congenital alloimmune hepatitis) presents a challenge in analysis and management [7].
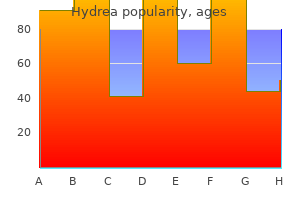
Buy cheap hydrea 500 mg line
All of the abnormalities described thus far have been autosomal recessive aside from X-linked adrenoleukodystrophy 3 medications that affect urinary elimination hydrea 500 mg purchase. One of the problems in trying to classify peroxisomal problems is that clinical syndromes have been linked to mutations in a variety of Table 37 treatment writing hydrea 500 mg cheap amex. Two types of classification are usually used: disorders of peroxisomal biogenesis and issues of single enzymes (Table 37. Those issues in which some sort of hepatic involvement has been reported are noted in Table 37. A temporary abstract of each of the peroxisomal issues for which hepatic disease has been described will be offered as will a dialogue of putative mechanisms underlying the liver disease, the method to making a biochemical diagnosis (Table 37. Zellweger syndrome is characterised by craniofacial abnormalities (wide anterior fontanelle, distinguished forehead, anteverted nostrils, low nasal bridge, epicanthal folds, flattened philtrum and narrow upper lip, along with bilateral clinodactyly and talipes equinus varus). Severe neurologic abnormalities are attribute, including hypotonia, areflexia, absent Moro response, profound intellectual incapacity, and seizures. Poor sucking is normally noted in the new child period and persists, resulting in severe failure to thrive. Skeletal radiographs demonstrate stippled epiphyses, and dislocated hips are common. Cerebral ventricles may be dilated, and cerebral atrophy with an irregular gyral sample is Group 2: single enzyme/protein deficiencies a Liver abnormalities. In normal human fetal liver, peroxisomes are present as early as the sixth week of gestation, but in children with Zellweger syndrome, hepatic peroxisomes are still absent at mid-gestation. Hepatic abnormalities embrace hepatomegaly (which may be slight and inconsistent or marked and persistent) and conjugated hyperbilirubinemia in early infancy, probably ensuing from intrahepatic biliary dysgenesis. Late within the first year of life, agency hepatomegaly with splenomegaly suggestive of cirrhosis and portal hypertension has been reported. Hepatic histology reveals extreme hepatic iron shops and a cholangiolar lesion characterised by tiny plugs of bile in the cholangioles, notably in the periportal space [32]. Electron microscopy of liver reveals absent peroxisomes and, occasionally, mitochondrial abnormalities. Liver histology revealed bile duct paucity, cholestasis, arterial hyperplasia, very small branches of the vena portae, and parenchymal destruction [33]. Accordingly, they administered cholic acid and chenodeoxycholic acid (each 100 mg/day) through the oral path to a 6-month-old boy with Zellweger syndrome. Biochemical indices of liver function improved as did the hepatic histology, coincident with a major lower in serum and urinary cholestanoic acids. A few residual peroxisomes may be seen in the liver, and hepatomegaly is attribute. An 11-year-old boy with intellectual incapacity and sensorineural deafness has been described in whom continual liver illness was thought of his major scientific drawback. The syndrome is characterised clinically by dysmorphic features in roughly 25%, massive anterior fontanelle, failure to thrive, feeding issues, and poor imaginative and prescient. Craniofacial abnormalities are milder than these in Zellweger syndrome and is most likely not noted until later in the first year of life. Reported abnormalities include round facies, flat occiput, high brow, frontal bossing, epicanthal folds, telecanthus, depressed nasal bridge, small mouth, protruding tongue, low-set ears, and quick neck. Hypotonia is current occasionally, though not as marked as in Zellweger syndrome, and peripheral reflexes are preserved. Sensorineural deafness (100%) and rotary nystagmus have been reported along with pigmentary retinopathy (Leber congenital amaurosis). Genitourinary abnormalities reported, embrace bilateral vesicopelvicalyceal dilation and vesicourethral reflux. Other hepatobiliary abnormalities embody isolated neonatal cholestasis without other organ system involvement. Cholelithiasis and mildly deranged liver function have been reported as early as 6 months of age. Liver illness may progress and turn into clinically important in children who survive the primary decade. Hepatic peroxisomes are absent or poor, and defects in bile acid metabolism are just like these characteristic of Zellweger syndrome. There is one report of profitable hepatocyte transplantation on this dysfunction [37]. Liver disease was not mentioned within the one report of sufferers with the isolated enzyme deficiency. Neonatal adrenoleukodystrophy Neonatal adrenoleukodystrophy can additionally be a defect of peroxisomal biogenesis. Approximately 25% of affected kids have some dysmorphic options; deafness is attribute, psychomotor delay is progressive, and hypotonia is reasonable as are seizures. The adrenals are small Group 2: isolated peroxisomal enzyme deficiencies D-bifunctional protein deficiency D-bifunctional protein contains each D -3-hydroxyacyl-CoA hydratase and D -3-hydroxyacyl-CoA dehydrogenase activities. Patients are hypotonic with mild craniofacial dysmorphism, multifocal tonic-clonic seizures, and calcific stippling of certain joints. Hepatomegaly and hepatic dysfunction have been reported; one affected person in our establishment additionally had liver fibrosis (G. Clinical features include profound hypotonia and dysmorphic options, including hypertelorism, epicanthal folds, low nasal bridge, low-set ears, and polydactyly. It is characterised by a steady, high urinary oxalate excretion and progressive bilateral oxalate urolithiasis and nephrocalcinosis. In the period before organ transplantation, death from renal failure occurred in childhood or early adulthood. Extrarenal deposits of calcium oxalate have been observed in skin, retina, and myocardium. We have noticed huge hepatomegaly secondary to calcium oxalate deposits in the liver of a 20-year-old patient with the dysfunction. Orthophosphate supplementation might forestall the development of calcium oxalate stones and small doses of a thiazide diuretic could additionally be helpful [38]. Since the primary enzyme defect is within the liver, renal transplantation is unsuccessful as a end result of the donor kidney is injured by steady deposits of calcium oxalate. One affected person suffered from livedo reticularis, peripheral gangrene, and third-degree heart block secondary to calcium oxalate sludge; all of those manifestations resolved following liver transplantation. Kidney plus liver had higher kidney graft outcomes than kidney alone, with death-censored graft survival of 95% versus 56% at 3 years (p � zero. Whether simultaneous liverΫidney transplant is completed (to provide maximal immunologic benefit) or sequential liver transplantation followed by kidney transplantation (which offers biochemical benefit) is dependent upon many elements together with disease staging, services, and entry to deceased or living donors [40]. Still one other reported approach is sequential liver and kidney transplants from the same living donor [43]. Novel non-transplant approaches embody restoration of faulty enzymatic exercise through the usage of chemical chaperones and hepatocyte cell transplantation, or recombinant gene remedy for enzyme alternative [45]. However, all the above non-transplant therapeutic approaches stay experimental at the present time. In sufferers with the disorder, enzyme activity in liver ranged from 11 to 47% of control values; the diploma of deficiency seems to be related to medical severity and the quantity of biochemical derangement.
Indian Root (American Spikenard). Hydrea.
- Colds, coughs, asthma, arthritis, skin diseases, promoting sweating, and other conditions.
- Dosing considerations for American Spikenard.
- What is American Spikenard?
- How does American Spikenard work?
- Are there safety concerns?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96383

500 mg hydrea generic with visa
Biopsy findings are variable however might embody areas of focal necrosis encircled by lymphocytes medications you cannot eat grapefruit with purchase 500 mg hydrea fast delivery, neutrophils medications diabetic neuropathy purchase 500 mg hydrea free shipping, and eosinophils. Resolution is usually full following enough remedy of the underlying infection. Patients might have mild elevation of serum aminotransferases; jaundice may occur. Treatment with doxycycline, erythromycin, and penicillin may be efficient, particularly if the analysis is made early in the disease course. Leptospirosis Leptospirosis is brought on by certainly one of several serotypes of Leptospira interrogans, a coiled, motile spirochete whose main hosts embrace a variety of domestic and wild animals. At-risk individuals have traditionally included those uncovered to cattle, hogs, horses, and rats. Exposure to blood, different body fluids, or fluids contaminated by urine from affected animals may end in illness transmission to humans. After an incubation period of 4Ͳ0 days, certainly one of two basic illness patterns might happen. Approximately 905% of sufferers in grownup collection will remain anicteric and bear an initial phase of illness lasting 4 days, marked by the presence of spirochetes in the peripheral circulation and characterized by fever, anorexia, abdominal ache, conjunctival erythema, lymphadenopathy, rash, and muscle tenderness. Approximately 50% of patients bear a second interval of fever, often marked by meningeal involvement, hepatitis, and, sometimes, endocarditis and myocarditis. In 5ͱ0% of patients, there might be a more severe course, marked by significant jaundice, renal failure, hemorrhage, and vascular collapse, with death occurring in up to 40%. Hepatomegaly was seen in 5 of 9 hospitalized children with leptospirosis; acalculous cholecystitis was additionally noted in 5, serum bilirubin >1. More latest sequence have confirmed these differences in presentation between children and adults. Other irregular laboratory findings may embody elevated serum creatinine phosphokinase, leukocytosis, thrombocytopenia, and proteinuria. Serum prothrombin time may be elevated but usually normalizes in response to vitamin K administration. The pathophysiology of leptospirosis-associated hepatic illness stays unsure. In approximately 10% of sufferers, small foci of hepatocellular necrosis may be present. Diagnosis of leptospirosis could also be made by way of culture of blood or cerebrospinal fluid throughout early stages of sickness, and from urine subsequently. Although not diagnostic, darkfield examination of the urine may provide helpful data. Borreliosis Lyme illness Lyme illness is caused by Borrelia burgdorferi, a tick-borne spirochete. Acute signs and symptoms embody erythema chronicum migrans, fever, malaise, headache, stiff neck, arthralgias, myalgias, and lymphadenopathy. Hepatic involvement in humans has been described; 19ͳ7% might have irregular liver tests [50]. Symptoms according to hepatic dysfunction may be elicited and embrace nausea, vomiting, anorexia, and weight reduction. Liver biopsy in a single affected person revealed infiltration of sinusoids by neutrophils and mononuclear cells [51]. Microvesicular fats, Kupffer cell hyperplasia, ballooning hepatocytes, and elevated hepatocyte mitotic exercise had been also famous. Diagnosis of Lyme illness requires a excessive index of suspicion on the part of the investigating physician. Treatment of early disease is with doxycycline [24]; cefuroxime or amoxicillin may be utilized in kids beneath 9 years of age. Parenteral remedy with ceftriaxone or penicillin V may be required in these sufferers with extreme carditis, persistent arthritis, or meningitis. Borrelia recurrentis Patients with Borrelia recurrentis an infection are additionally known to have hepatic involvement. Detail of the spirochete coiling is somewhat obscured by the silver deposits on this silver impregnation-based stain (WarthinΓtarry). Fibrinoid ring lesion containing a big, central lipid droplet surrounded by few inflammatory cells and encircled by a ring of fibrinoid material (arrow). Treatment of affected people with parenteral penicillin appears most efficacious if initiated inside the first few days of sickness [24]; ceftriaxone, cefotaxime, and doxycycline are additionally of use. Rickettsial illness Rocky Mountain noticed fever, the clinical syndrome related to Rickettsia rickettsii an infection, is characterized by fever; petechial rash beginning peripherally, spreading to the trunk, and often involving the palms and soles; and headache. Hepatic involvement might happen; medical manifestations embrace hepatomegaly and, not often, jaundice. Pathologic modifications noted at autopsy have consisted of portal triaditis, portal vasculitis, and erythrophagocytosis. Rickettsial organisms may be present in portal blood vessels and/or sinusoidal lining cells [55]. Q fever Q fever is attributable to Coxiella burnetti, a proteobacteria, and is characterized by fever, headache, malaise, myalgia, and pneumonitis, though asymptomatic infection predominates in people. Transmission occurs largely by way of inhalation of the Coxiella organism; this mode of transmission differs from rickettsiae, with which Q fever has been traditionally associated [56]. Symptomatic an infection in people lasts 9ͱ6 days, although acute infection may last as lengthy as three months. Chronic infection might occur, primarily in the form of Q fever endocarditis and osteomyelitis. Specifically, 705% of sufferers are noted to have abnormal liver checks, and 11Ͷ5% are famous to have symptoms referable to hepatic involvement [57]. In one middle, 42% of sufferers with Q fever presented with hepatitis in the absence of pulmonary signs [57]; 5% of sufferers present with jaundice. Although uncommon, hepatic failure secondary to Q fever has been documented in children. Early lesions might comprise neutrophils, whereas giant cells are noted in later lesions [58]. Non-specific modifications embody steatosis, mononuclear infiltration of portal areas, and Kupffer cell hyperplasia. Treatment is often problematic due to the self-limited nature of most infections; nevertheless, doxycycline is efficacious; co-trimoxazole is beneficial for kids youthful than 8 years of age. Incidence may be higher in particular teams, together with residents of group properties, male homosexuals, and immigrants from endemic areas. After traversing the stomach, ingested cysts dissolve during passage by way of the small bowel and colon where, in the presence of colonic bacteria, they mature into trophozoites. Colonic an infection may be asymptomatic or could manifest as invasive illness characterized by belly pain, bloody diarrhea, and the presence of "pipe stem" ulcers.
Order 500 mg hydrea with mastercard
A contracted and fibrotic quadriceps could result in a knee extension contracture medications in checked baggage discount hydrea 500 mg line, and in such cases quadricepsplasty is indicated along with the patella realignment symptoms 9 days after iui 500 mg hydrea mastercard. More generally, an related kneeflexion deformity could require hamstring release and posterior capsulotomy, though results have been inconsistent (163). Residual deformity, which is usually associated to flexion or rotation, is managed by femoral osteotomy towards the tip of the first decade of life. An intra-articular septum makes arthroscopic management difficult, but the septum may be eliminated arthroscopically. The radial head dislocation is asymptomatic in younger children, however may turn out to be symptomatic with time. Dislocated hips (173) and clubfeet can occur, and could be managed utilizing techniques just like these in idiopathic circumstances. There is nice variability within the age at onset and severity of the nephropathy (174). All patients should be referred for a nephrology analysis when this diagnosis is made. Patients could go on to persistent renal failure, requiring long-term nephrology management. The association of anomalies in the eye, ear, and vertebrae are termed ocularΡuricularζertebral dysplasia or Goldenhar syndrome (175). The traditional quartet of features consists of dystrophic nails (A), absent patellae (notice the region of osteochondritis dissecans on the lateral film) (B), posterior dislocation of the radial head (C), and iliac horns (D). It has an estimated incidence of 1 in 5600 births (178), and roughly 2% of individuals with congenital spinal abnormalities could have one other manifestations of ocularΡuricularζertebral dysplasia (138). Neural tube defect occurs extra typically than within the common inhabitants, and it could contain any portion of the spine, and even the cranium (an encephalocele). The congenital curve can cause cosmetic concerns, however these must be thought-about in the context of the other abnormalities, which may outweigh the beauty implications of the spinal deformity. In addition, Sprengel deformity and rib anomalies may be present in association with the congenital curves within the cervicalδhoracic area, and these contribute to the cosmetic implications of the situation. The congenital curves ought to be managed like congenital scoliosis of other etiologies, although management based mostly on cosmetic concerns needs to be made in the context of the opposite deformities. C: X-ray movie demonstrates the congenital anomalies of the lower cervical and the higher thoracic spine. Hypoplasia of the ascending ramus of the mandible accounts for the facial asymmetry. The compensatory curve can cause as a lot, if not more of a problem for the patient because the congenital curve. Brace therapy has no impact on the congenital curve, and though orthotic management has been used for the compensatory curve, its success fee appears decrease than for idiopathic scoliosis although high-quality comparative studies of its efficacy are missing. Intubation for anesthesia may be troublesome due to the small jaw, stiff neck, and higher airway dysmorphology (180). Mental retardation, reported to have an result on between 10% and 39% of sufferers, is more common in circumstances involving microphthalmia or an encephalocele (143, 182). Cornelia de Lange syndrome is related to a characteristic face, and growth retardation, which makes the clinical prognosis of Cornelia de Lange syndrome moderately reliable (183). Mutations in numerous genes, which all regulate the same signaling pathway, are identified in Cornelia de Lange syndrome. The mutation alters the activity of a developmentally essential signaling pathway known as Notch (186, 187). Notch plays a serious position in central nervous system growth, hence the related psychological retardation. Duplication or deletion of the chromosome band 3q25-29 produces a phenotype just like Cornelia de Lange syndrome (188, 189). In these cases, the mother is at all times the transmitting father or mother, suggesting genomic imprinting. They type a curious constellation of a small hand, a proximally positioned thumb, clinodactyly of the small finger, and decreased elbow movement, often caused by a dislocated radial head. These may be managed equally to cerebral palsy, however there seems to be a higher price of recurrence (198). There is presumably a higher incidence of Legg-Perthes illness, approaching about 10%. Scoliosis can occur and must be managed equally to scoliosis in cerebral palsy. The mortality price within the first yr of life is high because of faulty swallowing mechanisms (199), gastroesophageal reflux (200), aspiration, and respiratory infections. If the children survive their first year, they normally do nicely, but the long-term end result is unclear. Notice the traditional facial options of heavy eyebrows assembly in the midline, upturned nostril, downturned corners of the mouth, and lengthy eyelashes in a 13-year-old boy (A) and a 7-year-old girl (B). Cornelia de Lange syndrome: a baby with a severely affected higher extremity on her proper facet. There is retarded mentation, but the added options of no speech and no interactions cause main disability (201). Orthopaedic interventions must be thought of in the general practical context of the person. Braces, bodily remedy, and surgical procedure for tight heel cords, utilizing related indications as in cerebral palsy are justifiable. Patients with Cornelia de Lange syndrome rarely if ever use upper extremity prostheses. Lower extremity prostheses, nonetheless, ought to be prescribed for the uncommon case with tibial deficiency. Many teratogenic agents modulate the identical pathways which might be dysregulated by the mutations that trigger such syndromes. This may be attributable to mutations in a gene known as sonic hedgehog, and can also be brought on by teratogenic brokers that block the hedgehog signaling pathway, such as derivatives discovered within the plant Veraculum californium (205, 206). Fetal alcohol syndrome is a pattern of malformations found in kids of alcoholic mothers. There is a great deal of variability in the findings related to fetal alcohol exposure and the full-blown syndrome is normally seen only in youngsters of persistent alcoholics who drink all through pregnancy. Multiple phrases are used to describe the effects that outcome from prenatal publicity to alcohol, including fetal alcohol effects, alcohol-related delivery defects, alcoholrelated neurodevelopment disorder, and, most lately, fetal alcohol spectrum dysfunction (207). The total incidence of full-blown fetal alcohol syndrome is reported to be between 0. Their smallness and a loss of fat counsel a seek for endocrine dysfunction; the sufferers often look just like those who are deficient in progress hormone. Children with fetal alcohol syndrome present with a analysis of cerebral palsy clinics. Because of the variety of medical options, a joint consensus conference sponsored by the Centers for Disease Control advised that a analysis of fetal alcohol spectrum disorder requires all three of the characteristic dysmorphic facial features (smooth philtrum, skinny vermillion border, and small palpebral fissures), prenatal or postnatal growth deficit in top or weight, and a central nervous system abnormality. The 3-year-old patient is small and has the characteristic face of fetal alcohol syndrome.
Cheap 500 mg hydrea with mastercard
The impingement might cause labral tears and early cartilage injury that are probably to medications vertigo discount hydrea 500 mg gradually progress and limit her ability to train and play soccer medicine 666 colds hydrea 500 mg buy discount on-line. Finally, the situation of the tumor is extraordinarily essential as a quantity of tumors seem only in sure locations (Table 4-6). Bone tumors that occur in the posterior elements of the spine are mostly osteoid osteomas, osteoblastomas, and aneurysmal bone cysts, whereas bone tumors that occur in the anterior elements are most commonly eosinophilic granulomas. In the appendicular skeleton, the most typical epiphyseal tumors are chondroblastomas and aneurysmal bone cysts, whereas the most typical diaphyseal tumors are fibrous dysplasias and eosinophilic granulomas. The commonest metaphyseal tumors are nonossifying fibromas and unicameral bone cysts. Some bone tumors, including fibrous dysplasia, enchondromas, nonossifying fibromas, and osteochondromas, might current with multiple lesions. A affected person presenting with a single bony osteochondroma or exostosis should have a careful evaluate of the household historical past to determine if other members of the family had related bumps, suggesting the analysis of a number of hereditary exostosis, an autosomal dominant condition. If the bodily examination detects different osteochondromas, a cautious examination could detect angular deformity at wrists (ulnar deviation) and valgus deformity on the ankles secondary to tethering by the osteochondromas. This boy has multiple caf鮡u-lait spots with clean borders just like the coast of California. Most sufferers with type 1 neurofibromatosis may have 5 or extra caf鮡u-lait spots which are >1. Pigmented skin lesions termed caf鮡u-lait spots may be very useful within the differential diagnosis. These patients may have axillary freckling, and patients with kind 1 neurofibromatosis will often have cutaneous neurofibromas. Most patients with type 1 neurofibromatosis could have five or more caf鮡u-lait spots which are larger than 1. The skin can be inspected for any overlying changes similar to erythema or vascular engorgement from hyperemia. These findings are often seen in affiliation with main bone sarcomas such as an osteosarcoma. This boy has a number of caf鮡u-lait spots with rugged borders like the coast of Maine. Fair 4 Good Near normal chondroblastomas and eosinophilic granuloma, and malignant bone sarcomas, especially Ewing sarcoma, will typically have a large soft-tissue mass. The adjoining joint is thoroughly examined to examine for swelling, range of movement, and muscle atrophy to differentiate between pain secondary to the tumor and pain secondary to an intra-articular derangement. This affected person has a painful proximal tibial lesion with a large related soft-tissue mass measuring four by eight cm protruding posteromedially. These findings counsel an aggressive tumor similar to an osteosarcoma, so anteroposterior and lateral radiographs of the knee are recommended. He has been a aggressive swimmer since he was 6 years old and presently practices 6 days a week. If that had been the case, exercise modification, similar to cross-training, or correction of a easy coaching error may resolve his symptoms. On bodily examination, inspection of the shoulder reveals no muscle losing, swelling, or deformity. A affected person with imbalance of the rotator cuff muscular tissues could have impingement with tendonitis involving the supraspinatus tendon. Range of motion of the shoulders reveals elevation to 180 degrees bilaterally, external rotation with the arm at the side to 70 degrees bilaterally, and internal rotation to the point where the thumbs contact the spinous strategy of T4 on the proper and T9 on the left. The limited internal rotation on the left side indicates tight posterior constructions, a common discovering in patients who do overhead athletics. Muscle energy testing of shoulder flexion, abduction, inside, and external rotation are graded from 0 to 5, based on the scale of the Medical Research Council (Table 4-7) (47). Rotator cuff imbalance is usually seen in patients with weakness of the periscapular muscles, including the rhomboids, serratus anterior, subscapularis, and trapezius muscular tissues. A swimmer with shoulder pain might have ligamentous laxity with multidirectional instability, or rotator cuff tendonitis with impingement. Ligamentous laxity with instability may be evaluated by palpating glenohumeral translation. With the patient seated, the clinician evaluates the amount of glenohumeral translation by stabilizing the scapula and clavicle with one hand while pushing and pulling the proximal humerus in an anterior and posterior course with the other hand. Excessive laxity of the superior glenohumeral ligament will allow the humeral head to subluxate inferiorly. Ligamentous laxity of the left shoulder is evaluated with the patient seated, by holding the scapula and clavicle with the right hand whereas pushing the humeral head anteriorly and pulling it posteriorly with the left hand (arrows). The quantity of glenohumeral translation is measured in millimeters and compared with the uninjured shoulder. If the left shoulder is being examined, the clinician abducts the shoulder to ninety levels and gradually increases the quantity of exterior rotation utilizing the left hand. The clinician gently pushes the humeral head forward with the right thumb (arrow), whereas the fingers are strategically placed anteriorly in entrance of the humeral head, to forestall any sudden instability. Ligamentous laxity of the shoulder can be evaluated by applying a longitudinal inferior traction drive to the higher extremity whereas observing the gap between the humeral head and the acromion (arrow). Excessive laxity of the superior glenohumeral ligament will allow the humeral head to subluxate inferiorly; this phenomenon is termed a "sulcus sign. A swimmer with shoulder pain may have tendonitis involving the rotator cuff muscular tissues, notably the supraspinatus tendon. To decide if the affected person has tendonitis, the clinician performs impingement checks. If the patient has tendonitis involving the supraspinatus tendon, elevation of the arm to 180 levels, with the shoulder internally rotated, will cause discomfort as the infected tendon impinges against the anterior inferior acromion and coracoacromial ligament. Another method to detect impingement is to flex the shoulder forward to 90 levels in impartial rotation, with the elbow flexed to 90 degrees. A: the relocation test is a two-part check carried out with the shoulder kidnapped to 90 degrees. The clinician first performs a fear test and notes the quantity of shoulder exterior rotation when the affected person first experiences apprehension (arrow). B: the clinician then stabilizes the humeral head by pushing it posteriorly (downward arrow). If the affected person has tendonitis involving the supraspinatus tendon, elevation of the arm to 180 degrees with inner rotation of the shoulder will cause discomfort when the infected tendon impinges against the anterior inferior acromion and coracoacromial ligament (arrow). While supporting the arm with one hand, the clinician then pushes down on the forearm with the other hand (arrow), internally rotating the shoulder. Discomfort with this maneuver signifies impingement between the inflamed supraspinatus tendon and the anterior inferior acromion and the coracoacromial ligament. The labrum surrounds the glenoid cavity, deepening the glenohumeral joint, and the humeral head rests towards the labrum.
Buy 500 mg hydrea with visa
In mammals symptoms 5 weeks pregnant cramps 500 mg hydrea quality, failure to close the anterior neuropore results in anencephalia treatment venous stasis order hydrea 500 mg with visa, and the posterior neuropore in spina bifida. The means of neurulation is intimately linked to changes in cell form generated by the cytoskeleton (microtubules and microfilaments). Differential cell division seen in numerous areas of the neural plate would also contribute to the dimensions and shape of this region. In addition, those cells instantly adjoining to the notochord and those cells at the hinges of the neural groove may also help to mildew the neural tube. Separation of the neural tube from the ectoderm that may type the pores and skin requires adjustments in cell adhesiveness. These cells will migrate all through the embryo and will give rise to several cell populations. The chordamesoderm will generate the notochord, a transient organ whose features embrace inducing neural tube formation and establishing the physique axis. The dorsal (somitic) mesoderm will produce lots of the connective tissues of the body. The lateral plate mesoderm will give rise to the center, blood vessels, and blood cells, and the body lining cavities. Lastly, the head mesoderm will contribute to the connective tissues and muscles of the face. At the neural stage, the body plan has been established and the regions of the embryo that can kind limbs, eyes, heart, and the other organs have been determined. Each area has, nevertheless, appreciable capability for regulation, in order that if part of the area is eliminated a normal construction can still form. In later sections, limb and axial skeleton formation might be mentioned in more detail. It provides rise to the neurons and supporting glial cells of the sensory, sympathetic, and parasympathetic nervous techniques; the melanocytes of the dermis; and the cartilage and connective tissue elements of the pinnacle. Differences in adhesiveness between the anterior and posterior halves of the somites result in neural crest being prevented from migrating over the posterior halves. Thus, presumptive dorsal ganglia cells acquire adjacent to anterior halves, giving them a segmental arrangement. The brain of organized and specialised constructions from an initially quite simple group of cells. During growth, differences are generated between cells within the embryo that lead to spatial group, modifications in type, and the technology of different cell varieties. Each cell should act in accordance with the identical genetic instructions, however it must interpret them with regard to time and area. The key to understanding growth lies in cell biology, in the processes of signal transduction, and in the control of gene expression that end in adjustments of cell state, motion, and progress. The single most essential fact in development relies on the shocking finding that the developmental management genes are maintained via evolution. Thus, for many genes discovered in the invertebrate techniques, homologue genes have been identified in vertebrates they usually have comparable developmental roles in species starting from the fruit fly to fish to mouse to human. It is handy to distinguish three primary developmental processes, even though they overlap with, and influence, one another significantly. These are the emergence of pattern, cell differentiation, and change in form or morphogenesis. Failure to shut the posterior neuropore at day 27 ends in spina bifida, the severity of which depends upon how much of the spinal wire stays open. Pattern formation is the method by which spatial and temporal arrangements of cell actions are organized throughout the embryo in order that a well-defined structure develops. Pattern formation is crucial for the right improvement of every part of the organism. In the creating limb, for example, pattern formation allows the cells to know whether to make the higher arm or the fingers, and where the muscular tissues should type. Diagram of a transverse part by way of the trunk of an early 4-week (A) and late 4-week embryo (B). Sclerotome cells migrate from the somite, and these cells finally turn out to be chondrocytes. The capacity of cells to sense their relative positions inside a limited population of cells and to differentiate in accordance with this position has been the topic of intense analysis. Interestingly, sample formation in lots of methods has similar principles, and extra putting comparable genes. Many of the so-called homeotic genes that decide segment id in Drosophila have turned up in vertebrates and appear to play comparable roles in segmentation of structures such as the brain or the vertebral column. Homeotic genes are like embryonic switches, analogous to switches of railroad yards that directed trains into one path rather than one other. The name comes from the truth that mutations in a few of these genes lead to what known as a homeotic transformation, by which one body structure replaces one other. For example, in mice in whom Hoxd11 is mutated, anterior sacral vertebrae are reworked into lumbar vertebrae. Homeotic genes in all systems work equally: they code for proteins called transcription elements that control gene expression. What is extra, these molecules and pathways have been conserved over the course of evolution. Morphogenesis relies on a somewhat restricted variety of mobile activities and encompasses the formation of all tissues and organs from the primary embryonic tissue layers to the completed limb, spine, or brain. Those early steps take place in the "control room" for growth, and morphogenesis is then what occurs on the "factory flooring" - the actual meeting of the tissues and organs that make up the organism. In addition, spatial patterns of cell proliferation, folding of cell groups, rearrangement of cells, and cell migration make essential contributions to morphogenesis, the process that shapes the embryo. Finally, as the embryo develops, cells turn into completely different, and this course of culminates in the specialization of cells for specific features. Therefore, during improvement, morphogenesis give rise to structures appropriate to their position within the embryo and, within these structures, the differentiation of individual cells and their interactions are spatially ordered. Cell differentiation is the method during which cells turn into structurally and functionally different from one another, ending up as distinct types as muscle, bone, or cartilage. Since every cell of the organism has the identical genetic materials, the achievement and persistence of the differentiation state is dependent upon a series of signals that in the end control the transcription of particular genes. In people, the zygote gives rise to about 250 clearly distinguishable kinds of cells. One of the main targets of developmental biology is to discover how these differences emerge from the fertilized oocyte. In any organism, differentiation results in the manufacturing of a finite number of discrete kinds of cells, each with its peculiar repertory of biochemical actions and potential morphological configurations.
Buy hydrea 500 mg cheap
The most frequently used classification system to objectively quantify clubfoot rigidity is the Dimeglio clubfoot rating (11) symptoms nausea 500 mg hydrea trusted. The clinician discusses the natural history of the congenital clubfoot deformity as well as the present treatment and recommends that stretching and therapy should begin preferably within the subsequent few weeks symptoms right after conception proven 500 mg hydrea. The flexibility of the metatarsus adductus could be assessed by putting the thumb of 1 hand on the calcaneocuboid joint laterally and abducting the forefoot with the other hand. The family first suspected a problem when he was 4 months old and was still having issue holding his head up. He developed a seizure disorder at 1 12 months of age, and his seizures are now beneath good control with medicine. This affected person has developmental delay so the standard bodily examination may also embrace an in depth neurologic examination and developmental evaluation. The clinician grasps his hands, steadily pulling him into the sitting place, while on the lookout for head and trunk control. A youngster will normally have head control by 2 to four months of age and trunk control by 6 to 8 months of age (Table 4-1). In children, there are a collection of primitive reflexes, together with the Moro, grasp, neck-righting, symmetric tonic neck, and uneven tonic neck reflexes, that are present at delivery and then steadily disappear with with other neuromuscular problems, such as arthrogryposis and myelomeningocele, and is usually much less aware of nonoperative administration. The atypical clubfoot can be skinny or fats and are incessantly stiff, short, and chubby and with a deep crease on the plantar surface of the foot and behind the ankle. They could have shortening of the primary metatarsal with hyperextension of the metatarsophalangeal joint reflecting a plantarflexed first ray. If these reflexes persist beyond 10 months of age, it may be a sign of a neuromuscular dysfunction. The Moro reflex is elicited by gently lifting the infant with the right hand under the higher thoracic backbone and the left hand underneath the pinnacle. The toddler abducts the higher limbs, with spreading of the fingers, followed by an embrace. Similarly, extension of the neck causes extension of the upper limbs and flexion of the decrease limbs. The uneven tonic neck reflex is elicited by turning the pinnacle to the facet, which causes extension of the upper and lower extremities on the side toward which the top is turned, and flexion of the higher and lower extremities on the other side. The extensor thrust, an irregular reflex, is elicited by holding the infant underneath the arms and touching the feet to the ground, which causes a rapid extension of all of the joints of the decrease limb, progressing from the feet to the trunk. A normal toddler will flex rather than prolong the joints of the lower extremities when placed in this position. These primitive reflexes have to resolve with progress and development earlier than the kid will be succesful of walk independently. There are other primitive reflexes that progressively disappear in normal youngsters at different stages of improvement, together with the rooting, startle, Gallant, and Landau reflexes. The rooting reflex is elicited by touching the nook of the mouth, which causes the mouth and tongue to flip toward the aspect that was stimulated. The startle reflex is elicited by making a loud noise, which causes a mass myoclonic response resembling a Moro reflex, except that the elbows remain flexed. The Gallant reflex is elicited by stroking the side of the trunk, which causes the infant to bend the spine toward the facet that was stimulated, making a scoliosis convex to the opposite side that was stimulated. The Landau reflex is elicited by supporting the infant by the trunk in the horizontal susceptible position; the typical response is extension of the neck, spine, and extremities. The reflex is constructive if the toddler extends the upper extremities to break the autumn. The footplacement reaction is elicited by holding the toddler under the arms, then gently lifting the infant so that the dorsum of the foot or the anterior surface of the tibia touches the facet of the table. The foot-placement reaction often develops early in infancy and may persist until the age of 3 or 4 years. Bleck (12) evaluated 73 youngsters who were 12 months of age or older and had been still not but strolling to determine their prognosis for walking. One point was assigned for each primitive reflex that was nonetheless present, and one level was assigned for every postural reflex that was nonetheless absent (Table 4-4). A rating of two factors or extra indicated a poor prognosis for strolling, a one-point rating indicated a guarded prognosis, and a zero-point score indicated a great prognosis. The bodily examination continues by evaluating the spine for any scoliosis or kyphosis. The higher and lower extremities are examined to assess vary of movement and to document any contractures. If a contracture is recognized, the clinician attempts to passively right it to determine whether it is flexible or inflexible. The foot-placement response is elicited by gently lifting the infant in order that the dorsum of the foot or the anterior floor of the tibia touches the side of the table. The parachute reflex is elicited by holding the infant in the air within the prone position, then abruptly lowering the toddler headfirst towards the table, simulating a fall. The reflex is positive if the toddler extends the higher extremities as if to break the autumn (arrow). Primitive reflex Asymmetric tonic neck Neck righting Moro Symmetric tonic neck Extensor thrust Postural reflex Parachute Foot placement Prognosis for walking: 2 factors, poor; 1 level, guarded (might walk); 0 factors, good. If the athetosis is of the strain kind, it can often be "shaken out" of the limb by the clinician. The reflexes are also examined to determine if the patient has hyperreflexia, clonus, and a positive Babinski reflex. A 3-Month-Old Boy Is Referred for Evaluation Because He Is Not Moving His Right Arm. Shortly after delivering a wholesome 5250 g (11 lb 9 oz) child boy, the mom was informed that the infant was not shifting his right arm. The pregnancy was regular, however the delivery was tough because of proper shoulder dystocia. The delivery staff needed to apply considerable traction on the head to ship the baby. At the 2-month appointment with the pediatrician, he was shifting his hand but at all times stored the upper extremity at his facet. After a pediatric orthopaedic historical past and bodily examination, the clinician focuses on a detailed examination of the higher extremities, evaluating the paralyzed right aspect with the uninvolved side. It is necessary to distinguish between a brachial plexus palsy (a traumatic paralysis involving the upper extremity) and a pseudoparalysis secondary to osteomyelitis of the proximal humerus, septic arthritis of the shoulder, or a birth fracture. The treatment for each of those circumstances is totally different, and a delay in remedy of osteomyelitis or septic arthritis may be devastating. An infant with osteomyelitis, septic arthritis, or a birth fracture will often have swelling on the website, whereas an infant with traumatic brachial plexus palsy will have no swelling in the extremity, however might have swelling in the neck.
